Phylogenetic and network methods
T-BAS Toolkit
Tree-Based Alignment Selector Toolkit
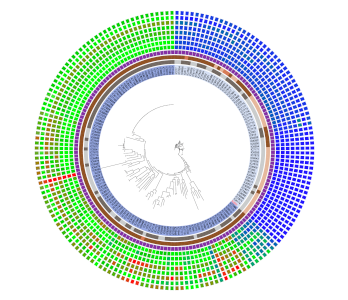
Patristic Distance
Create patristic distance rings with DendroPy

MAFFT
Align sequences with MAFFT

Tree Inference
Run de novo tree inference using RAxML
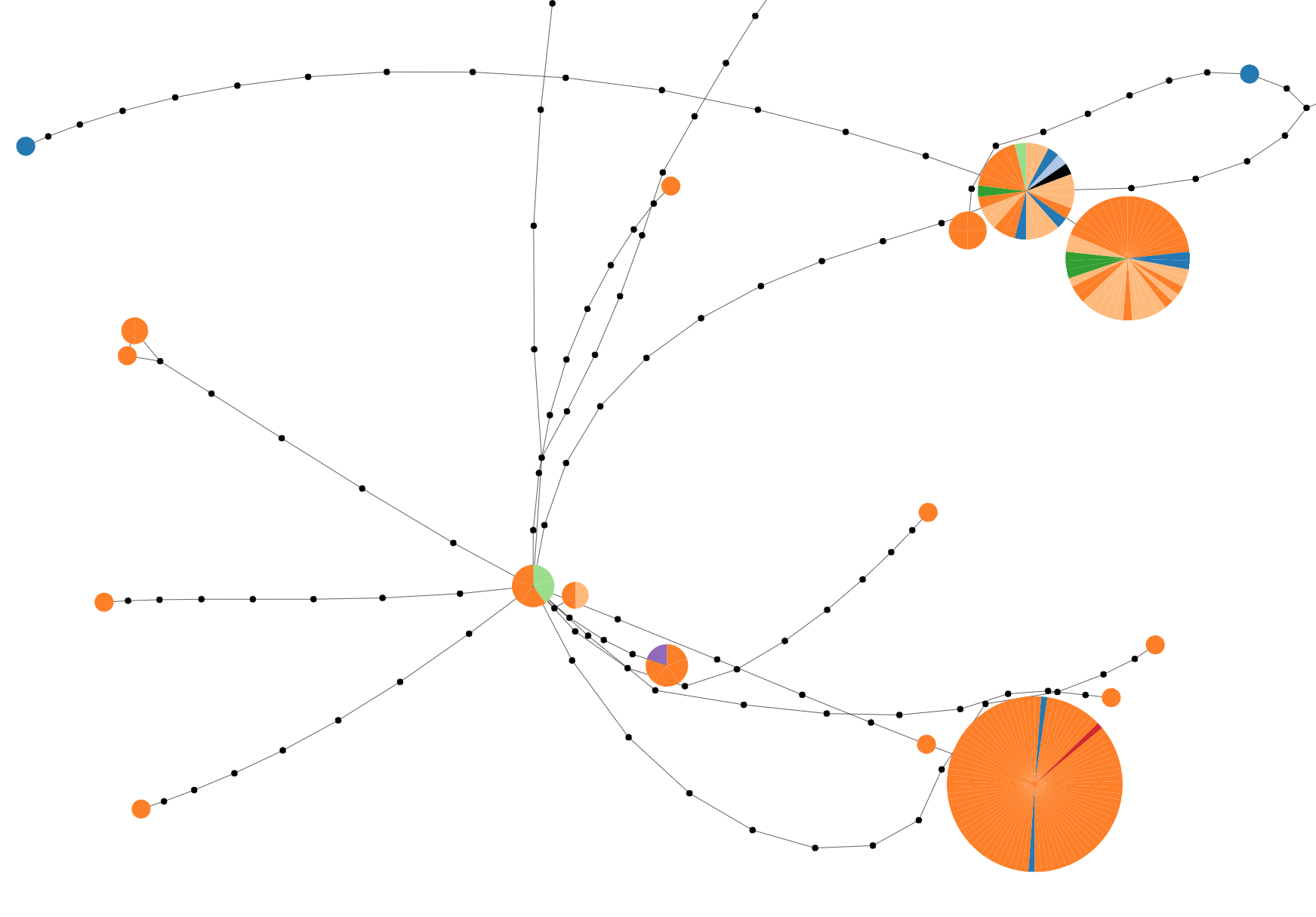
Graph Inference
Create networks using TCS or upload graph file

Mesquite Hypha
Show multiple node support values on tree using Mesquite Hypha
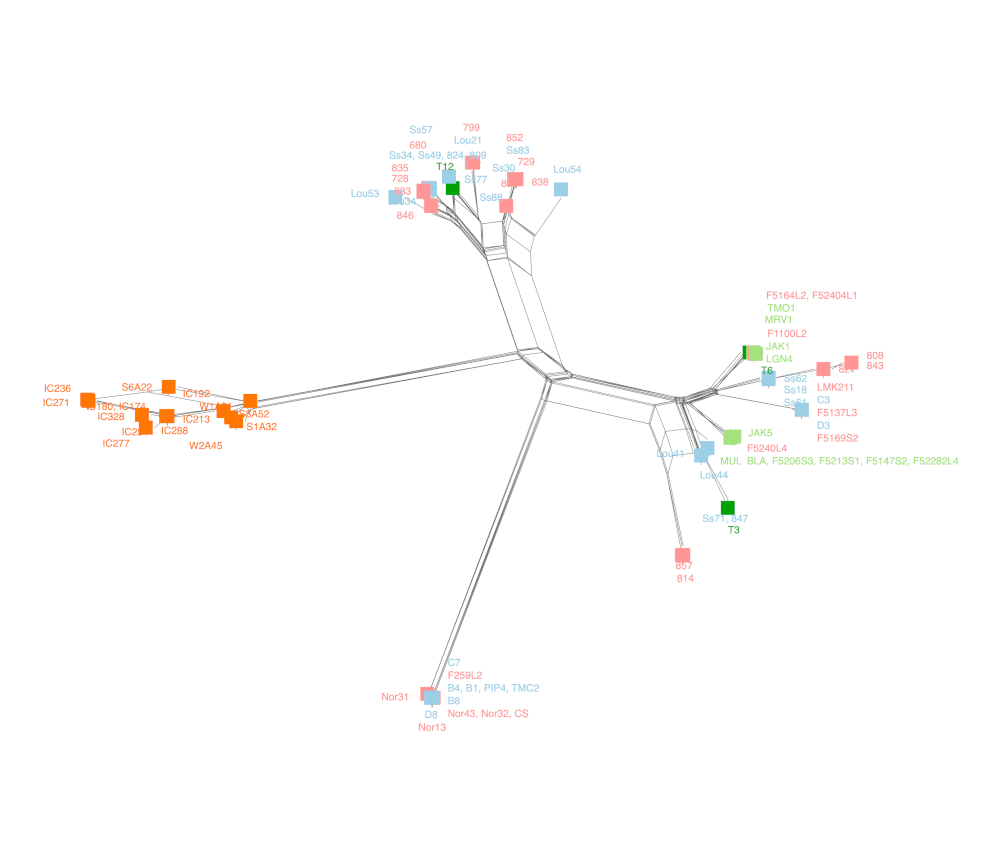
SplitsTree
Infer splits phylogenetic networks using Neighbor-Net
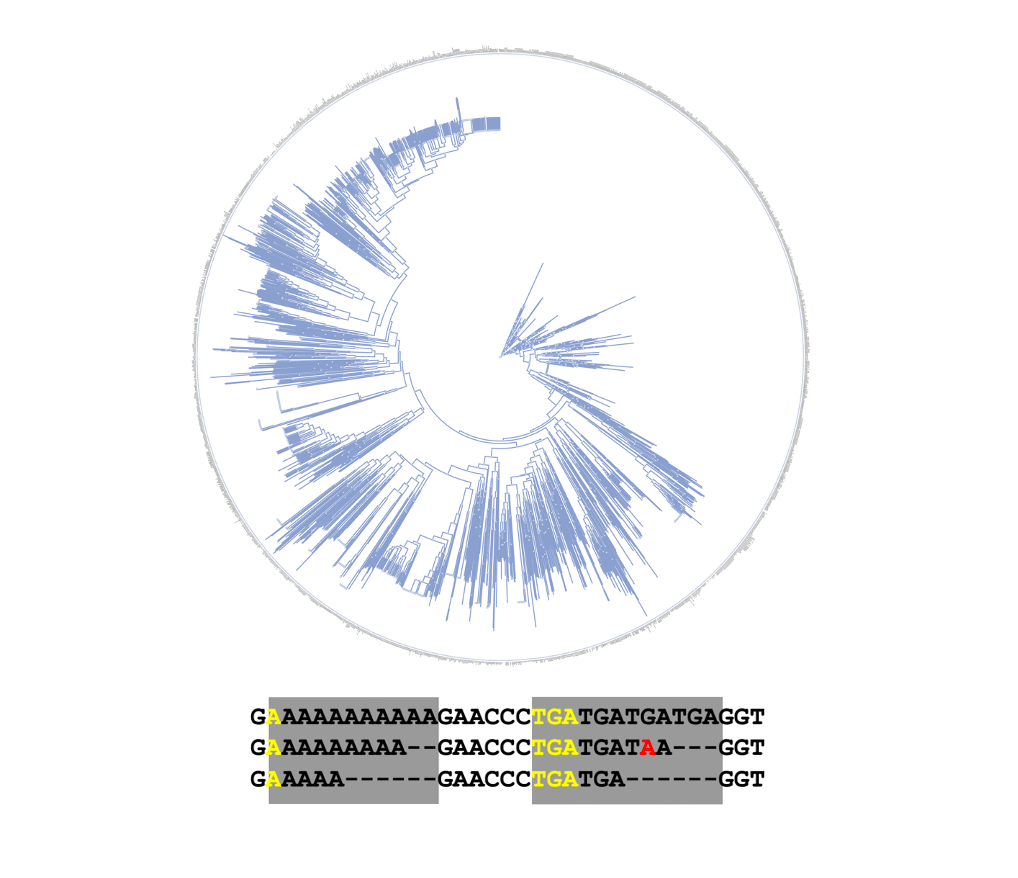
Microsatellite tree
Reconstruct tree from microsatellite data
Metabarcoding methods

MeShClust
Create haplotype table with MeShClust

Cluster Sequences
Cluster sequences with QIIME (UCLUST) or VSEARCH
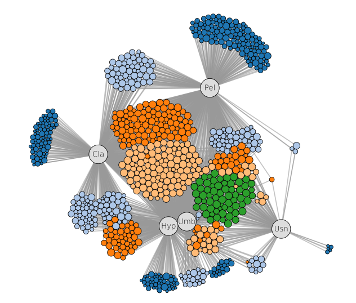
Graph CSV
Import csv file to graph

NGMLST
NGMLST: Next Generation Multilocus Sequence Typing using DADA2

DADA2
DADA2: Infer exact amplicon sequence variants (ASVs) from barcode sequences

Structure Files
Process data for structure tool

Phyloseq
Use phyloseq object to create graphs
Database search methods
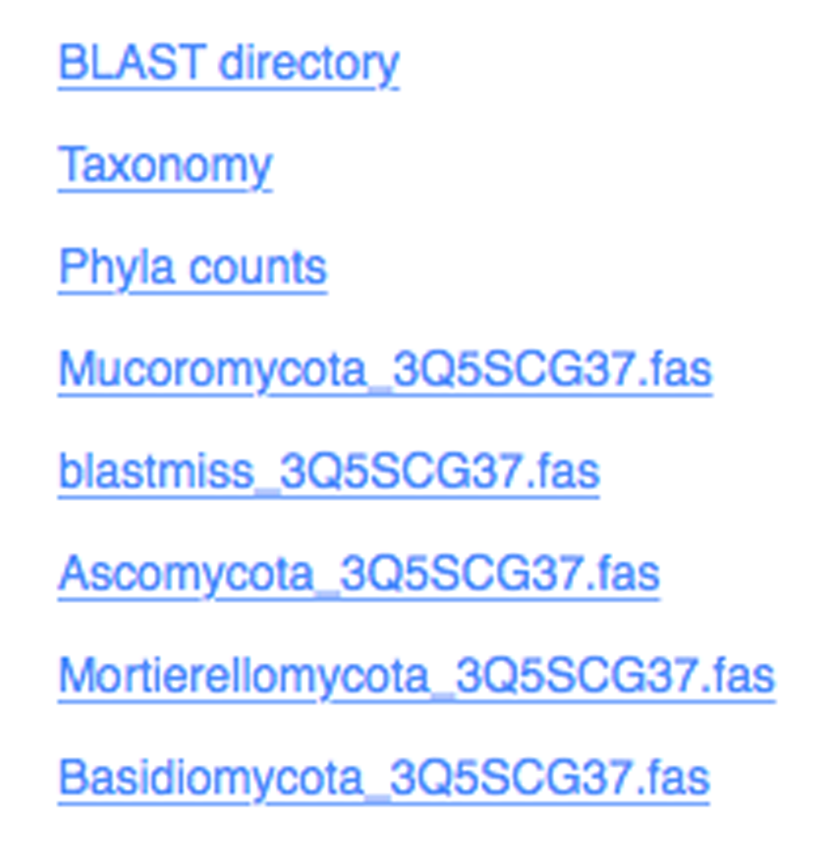
BLAST Taxonomy
Select fungal taxonomy using UNITE or NCBI ITS database

Query GenBank
Download FASTA and metadata for query of GenBank

ITSx
Split combined ITS/LSU into separate files with ITSx

Create Taxonomy
Create taxonomy csv file from list of scientific names

T-BAS Results Database
View T-BAS results from database for a run ID
T-BAS Specimen Database
View T-BAS results from database for specimens

Treebase
Download tree NEXUS files from treebase database

GNPS Library Search
Search in GNPS spectral libraries for metabolite

GNPS Ontology
Place MASST search results on NCBI taxonomy tree

GenBank IDs
Update T-BAS metadata with genus, species taxids.
File manipulation and conversion
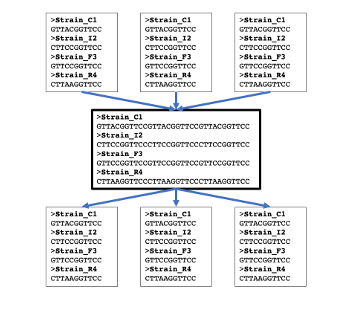
Concatenate Loci
Concatenate several loci or generate single locus files
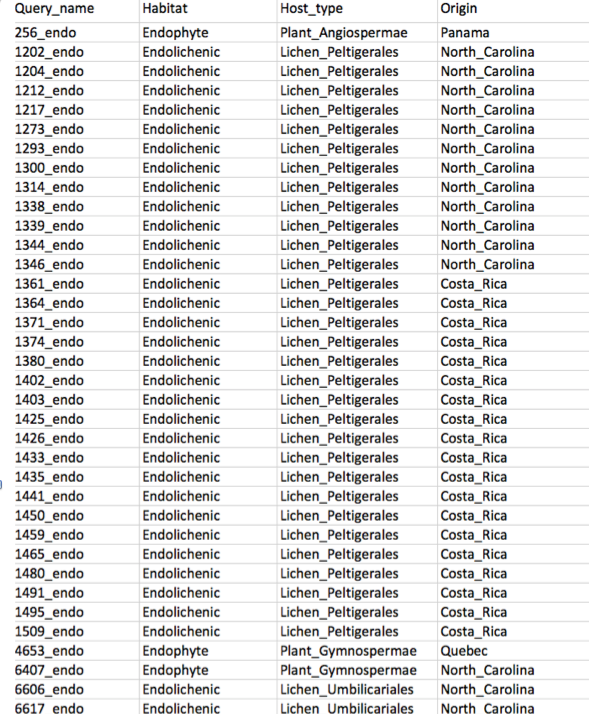
Merge Vouchers
Merge two voucher tables into one

Extend Sequence
Extend alignment with additional sequence
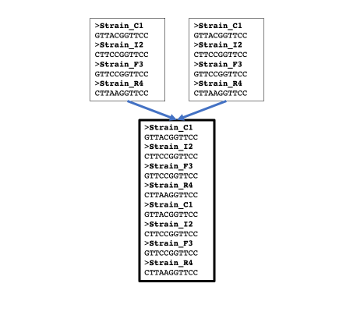
Append Files
Append several text files into one file
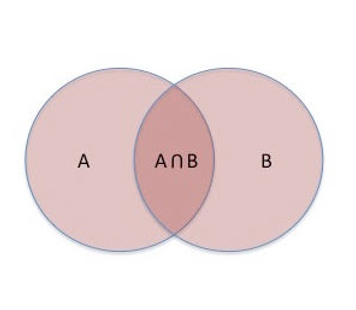
Get Set Results
Get set results from lists in 2 files

Nwk2nexus
Convert Phylogenetic tree from Newick to Nexus format
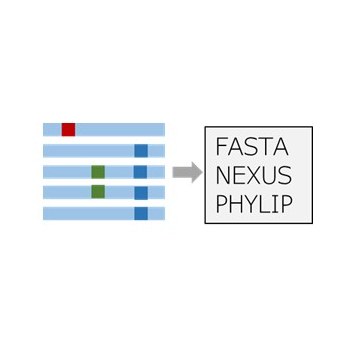
Sequence Converter
Convert sequence file(s) to different format with sites variable report
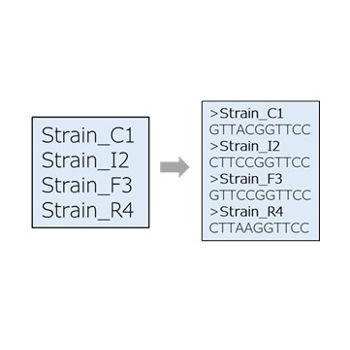
FASTA from list
Create new FASTA files using a strain list
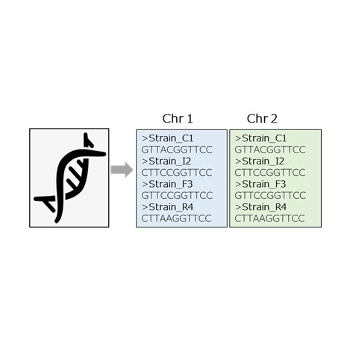
Extract Chromosome FASTA
Extract chromosome FASTA files from a Map file

Loci from Annotations
Create protein and DNA locus files from annotations using phmmer and nhmmer output, respectively
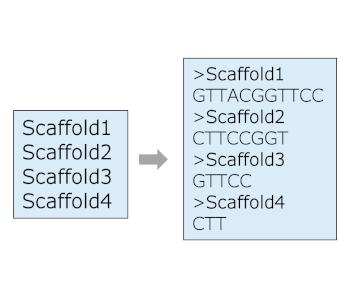
Sort FASTA
Sort FASTA file by lengths
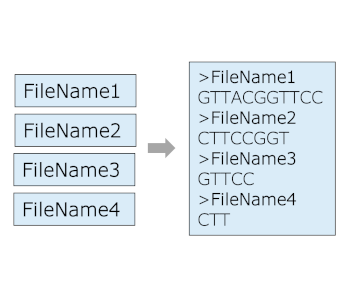
Rename FASTA headers
Rename FASTA headers with filename and append
Population genetic methods

Neutrality
Install locally and run neutrality test
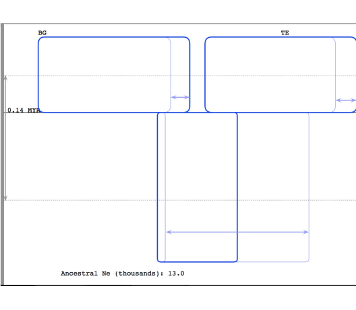
IMa3
Estimation of migration rates using Isolation-with-Migration (IMa3) models
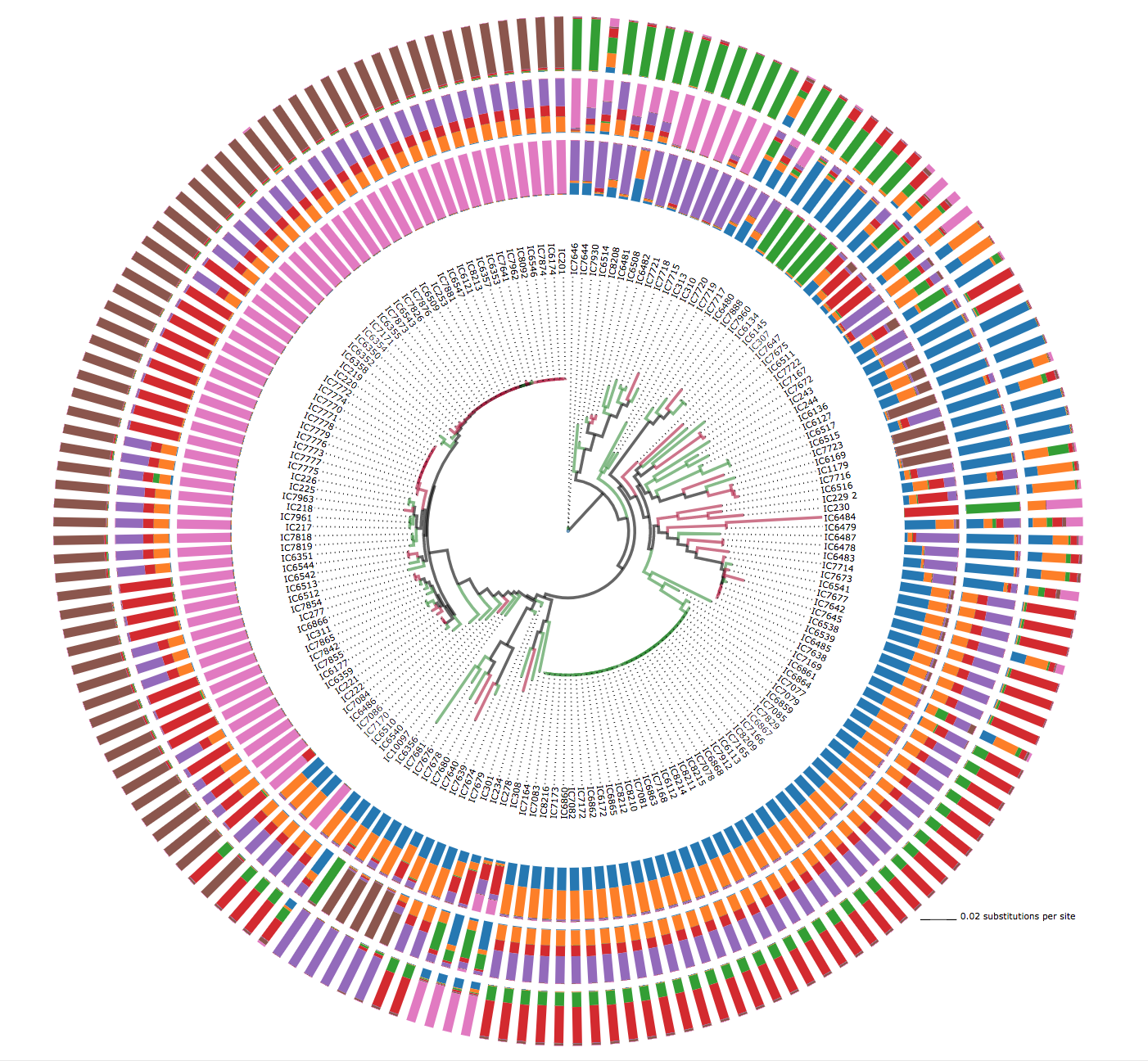
Structure
Show tree with structure plots

IMgc
IMgc on CIPRES
Galaxy@CIFR

Galaxy
Tools for population genetics and multi‑omics analysis

Galaxy - PPP
Popgen Pipeline Platform (PPP) for population genomic analyses

Galaxy - Cassava Virus Evolution
ViralSeq pipeline for NGS analysis of cassava mosaic begomoviruses
Utilities
Piechart
Create pie charts
Archive
Upload archive created by TBAS or DeCIFR utility
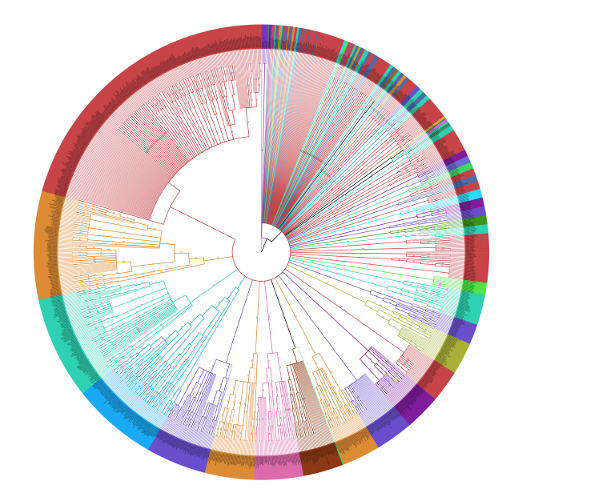
Collapse Tree
Collapse low support nodes on tree
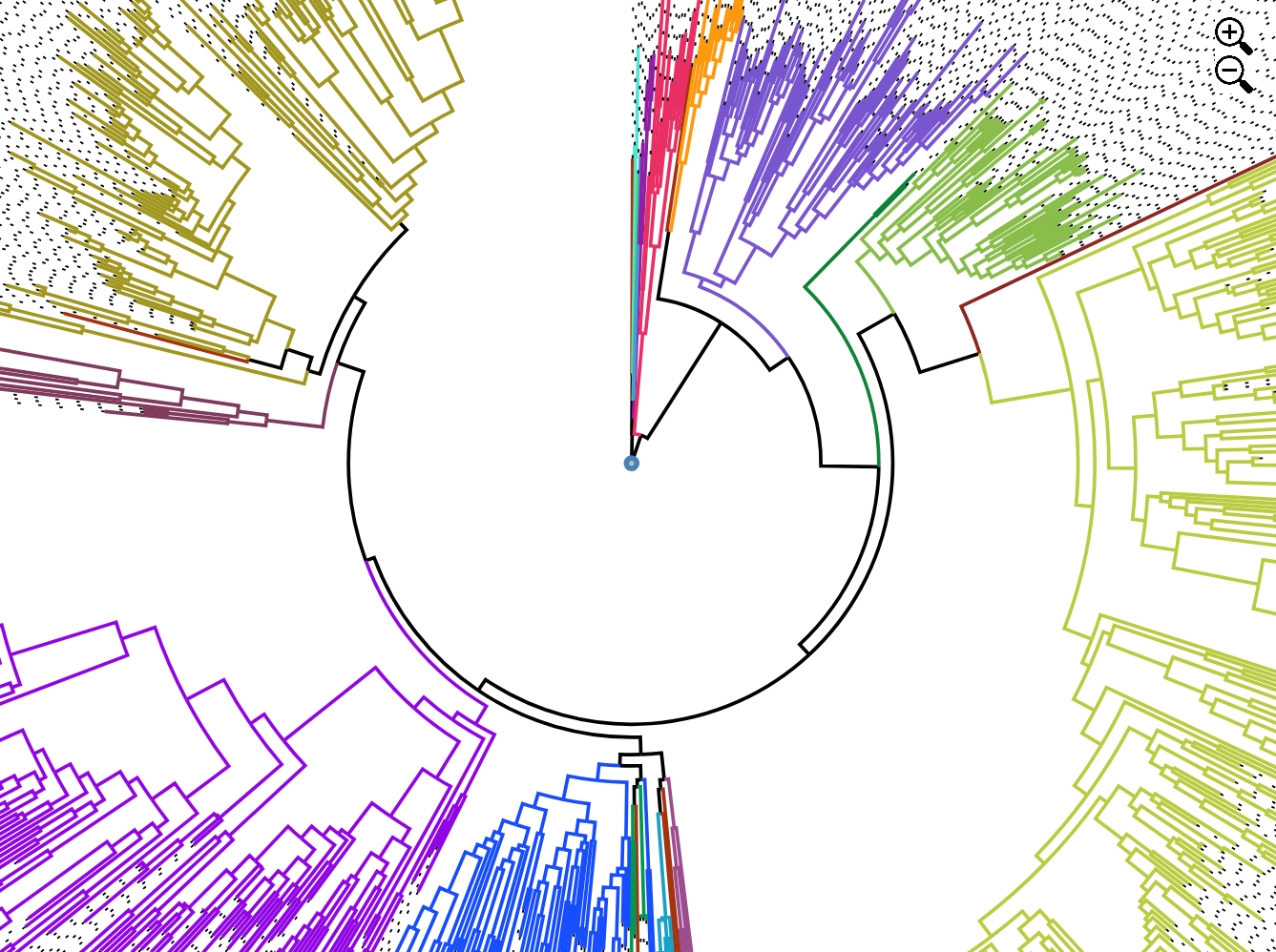
Telescope Viewer
View telescope tree from MEP file

MEP Archive
Retrieve MEP files from archive by run ID

Retrieve Names
Retrieve names from tree or sequence file
Update Names
Update names in tree or sequence file with replacements from csv

Extract Taxonomy
Create taxonomy CSV file from taxon string

Filter FASTA
Create FASTA locus files from genome annotations

CIPRES Jobs
Get CIPRES XML from jobs URL

CIPRES Jobs Database
Show jobs submitted to CIPRES (admin)

CIPRES Jobs Database User
Show jobs submitted to CIPRES (user)
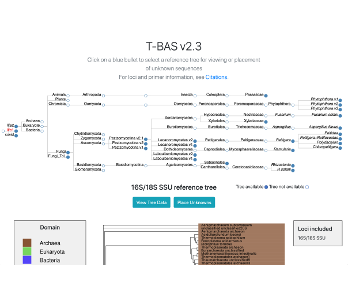
Guide Tree Edit Tool
Add new accessions to JSON config file for T-BAS guide tree

D3 SVG and Canvas
Big Tree Viewer

REST
Unzip MEP file for REST

T-BAS accession
Retrieve submitted tree, alignments, and metadata from archive by accession ID
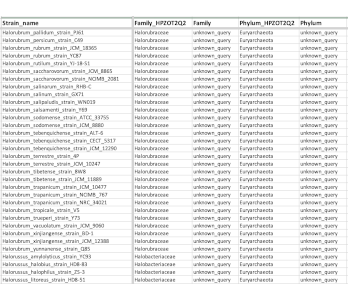
Merge Metadata
Merge columns in T-BAS metadata
PopPhy-CNN
Convolutional neural networks to predict host phenotype from metagenomic data
